
TL;DR:
- Researchers from the University of Eastern Finland, in collaboration with industry and supercomputers, achieved a 10-fold time reduction in processing 1.56 billion drug-like molecules using machine learning.
- Large compound libraries require rapid screening to identify potential drug agents.
- Machine learning, specifically the HASTEN tool, was used to predict docking scores for compounds, reducing screening time dramatically.
- This study demonstrates the efficacy of machine learning in virtual drug screening at an unprecedented scale.
- The authors have released the datasets generated during the study to the public, facilitating further advancements in computational drug discovery.
Main AI News:
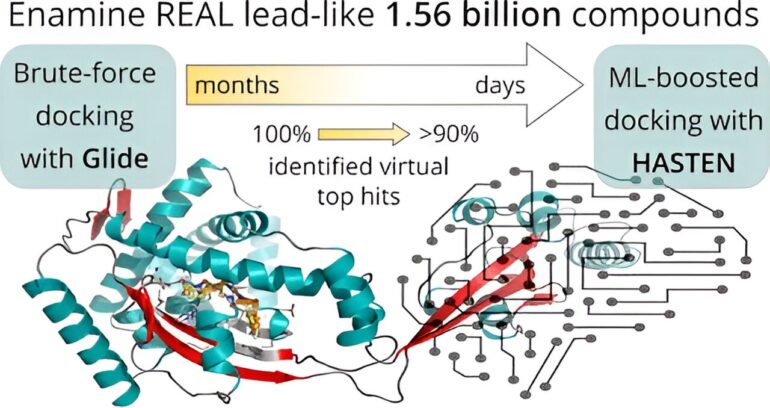
In the relentless pursuit of novel drug molecules, researchers face an ever-growing challenge—screening colossal compound libraries at a pace that matches the explosive expansion of these repositories. The University of Eastern Finland, in collaboration with industry leaders and the computational prowess of supercomputers, has achieved a breakthrough that promises to reshape the landscape of drug discovery. Harnessing the power of machine learning, they’ve achieved a staggering 10-fold reduction in screening time for a staggering 1.56 billion drug-like molecules. This remarkable feat is setting new standards in the world of pharmaceutical research.
In their quest to identify potent agents capable of blocking specific drug targets, researchers rely heavily on computer-aided screening. These targets may range from enzymes that bolster bacterial resistance to antibiotics to viruses seeking to infiltrate host cells. However, the exponential growth of compound libraries has left even state-of-the-art supercomputers struggling to keep pace.
The reality is that screening a billion-scale compound library against a single drug target can drag on for months, if not years. The urgency for swifter approaches is palpable, and this is precisely where machine learning enters the picture.
In a groundbreaking study published in the esteemed Journal of Chemical Information and Modeling, Dr. Ina Pöhner and her team from the University of Eastern Finland’s School of Pharmacy, in collaboration with CSC—IT Center for Science Ltd and Orion Pharma, embarked on a mission to unlock the potential of machine learning in turbocharging gargantuan virtual screens.
Before introducing artificial intelligence into the equation, the researchers laid down a baseline. In a monumental virtual screening campaign, they meticulously assessed 1.56 billion drug-like molecules against two pharmacologically relevant targets. This Herculean effort spanned nearly half a year and enlisted the computational prowess of supercomputers Mahti and Puhti, as well as molecular docking—a technique that fits small molecules into a target’s binding site, yielding a “docking score” as a measure of fit.
Subsequently, they compared their findings to the results generated by a machine learning-boosted screening tool named HASTEN, developed by Dr. Tuomo Kalliokoski from Orion Pharma and a co-author of the study.
“HASTEN harnesses the power of machine learning to discern the nuances of molecular properties and their impact on compound scoring. With enough data gleaned from traditional docking, the machine learning model swiftly predicts docking scores for other compounds in the library, bypassing the arduous brute-force docking approach,” elucidates Kalliokoski.
Astoundingly, with just 1% of the entire library docked and used as training data, HASTEN accurately identified 90% of the highest-scoring compounds in less than ten days.
This study represents the maiden comprehensive comparison between a machine learning-boosted docking tool and a conventional docking baseline on such a monumental scale. “Our findings unequivocally attest to the reliability and efficiency of the machine learning-boosted tool in expeditiously replicating the top-scoring compounds identified through conventional docking,” remarks Pöhner.
Professor Antti Poso, leading the computational drug discovery group within the University of Eastern Finland’s DrugTech Research Community, emphasizes the fruitful collaboration between academia and industry that made this endeavor possible. He underscores, “By amalgamating our ideas, resources, and cutting-edge technology, we’ve managed to achieve our ambitious objectives.”
Studies of this magnitude are a rarity, and in recognition of this, the authors have generously made the vast datasets generated during this study available to the public. Their comprehensive screening library for docking not only facilitates accelerated screening efforts but also serves as invaluable benchmarking data, featuring 1.56 billion compound-docking results for two pivotal drug targets.
This altruistic act is poised to spur the development of tools that optimize time and resources, propelling the field of computational drug discovery into an exciting new era. As the world grapples with ever-evolving health challenges, the fusion of machine learning and pharmaceutical science promises to be a potent force for good, unlocking innovative therapies at an unprecedented pace.
Conclusion:
The integration of machine learning into drug discovery, as exemplified by this study, signifies a major leap forward for the pharmaceutical industry. It not only accelerates the screening of vast compound libraries but also underscores the potential for AI-driven solutions to reshape drug development timelines. This innovation has the potential to expedite the introduction of novel therapies into the market, addressing health challenges more rapidly and efficiently. Pharmaceutical companies should take heed of this advancement and consider adopting similar AI-driven approaches to remain competitive in an increasingly dynamic market.