
TL;DR:
- Stanford’s GLOW and IVES protocols enhance molecular docking accuracy with deep learning.
- They outperform baseline methods, especially in dynamic binding pockets.
- Accurate ligand pose prediction is vital for drug discovery.
- GLOW and IVES offer curated datasets and open-source Python code.
- GLOW uses a softened van der Waals potential, while IVES incorporates multiple protein conformations.
- Both protocols excel in sampling ligand poses, even in complex scenarios.
- IVES benefits geometric deep learning on protein structures with fewer conformations.
- Datasets for 5,000 protein-ligand pairs are provided by GLOW and IVES.
Main AI News:
In the realm of molecular docking and ligand binding pose prediction, a groundbreaking transformation is underway. The potential of deep learning to elevate the accuracy of scoring functions cannot be overstated. However, existing sampling protocols often grapple with the need for prior information to yield precise ligand binding poses, thereby limiting the overall accuracy of scoring functions. Enter GLOW and IVES, two pioneering protocols engineered by the astute minds at Stanford University, poised to tackle this challenge head-on and usher in a new era of efficacy in pose sampling.
The Dynamic Challenge of Molecular Docking
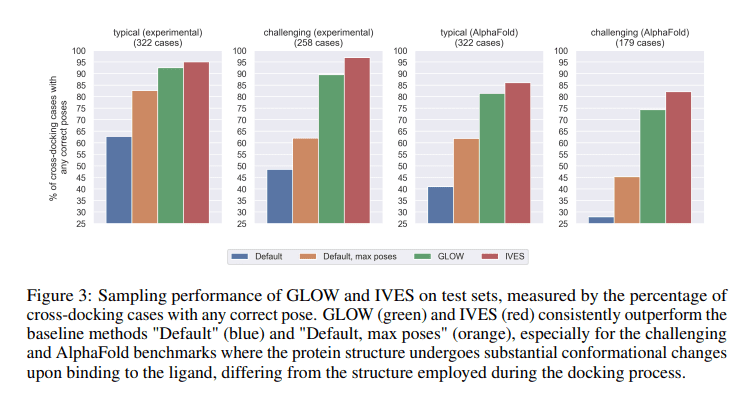
Traditional deep learning approaches to molecular docking typically hinge on rigid protein docking datasets, largely neglecting the inherent flexibility of proteins. While these methods are effective to a certain extent, they fall short in capturing the nuanced intricacies of protein flexibility. GLOW and IVES, however, stand as advanced sampling protocols that boldly address these limitations, consistently surpassing the performance of baseline methods. Their prowess shines particularly bright when dealing with dynamic binding pockets, opening the door to a promising future for the improvement of ligand pose sampling in protein-ligand docking—a critical facet of advancing deep learning-based scoring functions.
Empowering Drug Discovery through Accurate Ligand Prediction
At the heart of drug discovery lies the precise prediction of ligand placement within protein binding sites. Conventional methods, though valiant in their efforts, often stumble in generating accurate ligand poses. Deep learning emerges as a beacon of hope for enhancing accuracy in this domain, but its success is intrinsically tied to the quality of pose sampling. GLOW and IVES emerge as the torchbearers in this endeavor, augmenting the quality of samples in the face of challenging scenarios, thus elevating accuracy levels. Their versatility extends to both unliganded protein structures and those predicted, including those generated by AlphaFold, offering meticulously curated datasets and accessible Python code to all who seek to harness their potential.
The GLOW and IVES Paradigm
Let us delve deeper into the essence of GLOW and IVES, the two revolutionary pose sampling protocols that are reshaping the landscape of molecular docking. GLOW ingeniously employs a softened van der Waals potential to generate ligand poses—a strategy that has proven to be highly effective. In contrast, IVES takes accuracy to a whole new level by incorporating multiple protein conformations, unlocking a realm of possibilities.
A Resounding Victory over Baseline Methods
When the performance of GLOW and IVES is pitted against baseline methods, the results speak volumes. These two protocols consistently outshine their counterparts, especially when faced with the complexity of AlphaFold-generated protein structures. The evaluation of test sets serves as a resounding testament to their superior ability to sample correct postures. For IVES, the quality of the seed pose is paramount for efficient operation, with the Smina docking score and score being integral to the selection process.
Unleashing the Power of GLOW and IVES
GLOW and IVES, with their exceptional ability to accurately sample ligand poses, excel in even the most challenging scenarios and surpass AlphaFold benchmarks with ease, all while accommodating significant protein conformational changes. Additionally, IVES, with its knack for generating multiple protein conformations, serves as a boon for geometric deep learning on protein structures, achieving performance on par with Schrödinger IFD-MD but with fewer conformations required. To further fuel the advancement of deep-learning-based scoring functions in molecular docking, GLOW and IVES generously provide datasets comprising ligand pose information for a whopping 5,000 protein-ligand pairs.
Conclusion:
The introduction of GLOW and IVES by Stanford University marks a significant advancement in molecular docking and ligand binding pose prediction. These protocols promise to elevate accuracy and efficacy in the field, offering valuable tools for drug discovery and geometric deep learning. Their availability as open-source resources further fuels innovation and progress in the market.