
TL;DR:
- SPACEL, a Deep Learning-based toolkit, revolutionizes spatial transcriptomics (ST) analysis.
- Developed by Prof. Qu Kun’s team, it comprises three modules: Sprint, Splane, and Scube.
- Sprint accurately predicts cell-type distribution through innovative techniques.
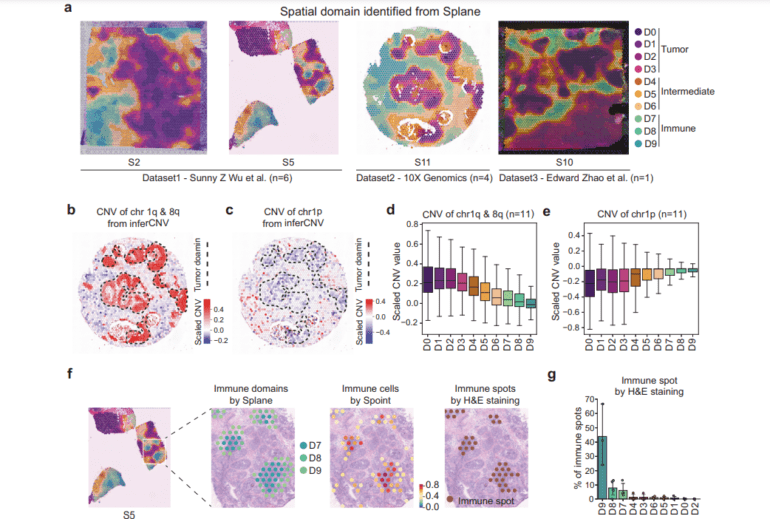
- Splane efficiently identifies spatial domains, eliminating batch effects in ST slices.
- Scube automates the alignment of slices, constructing a 3D tissue architecture.
- SPACEL outperforms previous techniques in cell type prediction, domain identification, and 3D reconstruction.
- Superior performance against 19 cutting-edge techniques on simulated and real ST datasets.
- SPACEL promises to reshape the landscape of ST data analysis.
Main AI News:
Spatial transcriptomics (ST) has long been a cornerstone of cellular research, enabling scientists to delve into the intricate world of gene expression within individual cells while preserving their spatial context. However, as the demand for more comprehensive insights into tissue organization grows, so do the challenges of analyzing multiple tissue slices simultaneously, hindered by the limitations of spot size in ST slices.
In response to these challenges, a pioneering team led by Professor Qu Kun from the University of Science and Technology of the Chinese Academy of Sciences has unveiled a groundbreaking solution: Spatial Architecture Characterization by Deep Learning, or SPACEL for short. This innovative toolkit comprises three modules – Spoint, Splane, and Scube – synergizing to construct a three-dimensional panorama of tissues automatically.
Sprint, the first module, tackles the intricate task of cell-type deconvolution. Leveraging a fusion of simulated pseudo-spots, neural network modeling, and statistical recovery of expression profiles, Sprint delivers pinpoint accurate predictions. Meanwhile, Splane, the second module, employs a graph convolutional network (GCN) approach and an ingenious adversarial learning algorithm to discern special domains. By concurrently analyzing multiple ST slices, Splane effectively eliminates batch effects and uses cell-type composition as its guiding input. A standout feature is its efficient identification of spatial domains. Lastly, Scube, the third module, streamlines the alignment of slices and constructs a stacked 3D tissue architecture, offering a comprehensive understanding of the tissue’s three-dimensional structure.
The true test of SPACEL’s mettle came as the researchers applied it to 11 ST datasets, encompassing a total of 156 slices, and engaged a repertoire of technologies such as 10X Visium, STARmap, MERFISH, Stereo-seq, and Spatial Transcriptomics. Remarkably, SPACEL surpassed previous techniques in three pivotal analytical tasks: cell type distribution prediction, spatial domain identification, and three-dimensional tissue reconstruction.
In addition, SPACEL demonstrated its supremacy in cell type deconvolution, spatial domain identification, and 3D alignment when pitted against 19 cutting-edge techniques, both in simulated and real ST datasets. With its superior performance and simplified approach, SPACEL emerges as the harbinger of precise understanding in the realm of ST data analysis. In the world of scientific inquiry, SPACEL is poised to revolutionize the way we explore the spatial transcriptomic landscape.
Conclusion:
SPACEL’s emergence signifies a major shift in the spatial transcriptomics analysis market. Its innovative approach to resolving challenges in ST data analysis, coupled with its superior performance over existing methods, positions it as a game-changer. Researchers and institutions seeking precise insights into cellular spatial organization and gene expression will likely gravitate towards SPACEL, potentially establishing it as the industry standard for ST analysis.