
- Century-long advancements in computing and high-throughput screening have driven the development of sophisticated molecular representations.
- Early chemical notations evolved to meet the demands of computational processing for digital storage and manipulation.
- AI applications leverage molecular graphs and other notations for efficient computational analysis in drug discovery.
- Contemporary formats like SMILES and InChI offer standardized, machine-readable representations for diverse molecular structures.
- Graphical representations, from 2D depictions to advanced 3D models, enhance visualization and analysis in cheminformatics.
Main AI News:
The past century has witnessed significant technological advancements, particularly in the realms of computing and high-throughput screening within drug discovery. These advancements necessitated the evolution of molecular representations that are not only comprehensible across scientific disciplines but also machine-readable. Initially, molecules were depicted in simple structure diagrams depicting bonds and atoms. However, the rise of computational processing demanded more sophisticated representations. Various chemical notations emerged to encode molecular structures. Early examples, such as the empirical formula, offered atomic composition without details on connectivity or geometry. The advent of computers enabled the rapid digital storage and manipulation of chemical data, leading to the development of machine-readable notations and algorithms supporting both 2D and 3D visualization. Modern representations, especially those developed post-1970s, encompass a wide array of forms capable of supporting small molecules, macromolecules, and chemical reactions, thereby enhancing the efficiency and scalability of cheminformatics.
Applications of AI in Drug Discovery
In the realm of AI-driven drug discovery, the role of chemical representations cannot be overstated. Molecular graphs, among the most prevalent machine-readable representations, are employed alongside various other notations to encode structural information for computational analysis. This review underscores the pivotal role played by these representations in AI applications. It provides concrete examples where AI techniques, such as machine learning models, are applied to cheminformatics and drug discovery. The review serves as an indispensable resource for researchers and students across chemistry, bioinformatics, and computer science disciplines, highlighting the criticality of selecting appropriate representations tailored to specific tasks. While not exhaustive, the review serves to guide readers towards further literature on AI applications within cheminformatics, demonstrating how modern computational techniques are revolutionizing drug discovery by bolstering data handling and analytical capabilities.
Introduction to Molecular Graph Representations
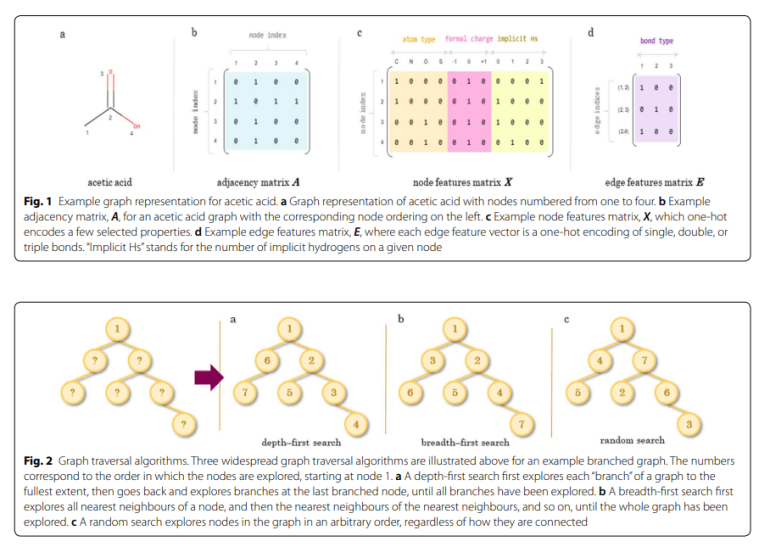
Understanding molecular graphs forms the bedrock for comprehending the chemical representations employed in drug discovery. These graphs map atoms to nodes and bonds to edges, systematically representing molecules in a structured manner. Formally defined as a tuple comprising nodes (atoms) and edges (bonds), these graphs are visualized using various software tools. Nodes and edges are typically encoded into matrices: an adjacency matrix for connectivity, a node features matrix detailing atom identities, and an edge features matrix specifying bond identities. The flexibility of these representations allows for the inclusion of 3D information, offering distinct advantages over linear notations.
Connection Tables and MDL File Formats
Connection tables (Ctabs) and MDL (now BIOVIA) file formats play indispensable roles in the representation of molecular graphs. Ctabs, structured into counts, atoms, bonds, atom lists, Stext, and properties blocks, efficiently describe molecular structures by detailing atom and bond specifics while omitting explicit hydrogen representations to reduce file sizes. MDL formats, built upon Ctabs, encompass Molfiles for individual molecules and extend to SD, RXN, RD, and RG files for additional data and reactions. Widely adopted for their compact and systematic approach to chemical information storage and transfer, these formats support diverse applications within cheminformatics.
Contemporary Notations: SMILES and InChI
SMILES, introduced in 1988, stands out as an intuitive and widely adopted notation for encoding molecular structures. Employing a depth-first search methodology, SMILES assigns numerical values to atoms and traverses molecular graphs to generate multiple representations of the same molecule, with unique SMILES designated through canonicalization. While adept at encoding stereochemistry and complex structures, SMILES encounters challenges with organometallic compounds and ionic salts. Conversely, the International Chemical Identifier (InChI), unveiled in 2006, offers a standardized, open-source canonical notation featuring multiple layers for comprehensive molecular representation. InChIKeys provide unique, searchable hashed versions of InChIs, thereby enhancing accessibility to chemical information.
Summary of Chemical Representations
Chemical representations encompass a myriad of methodologies for modeling molecules, reactions, and macromolecules. Structural keys such as MACCS and CATS encode the presence of specific chemical groups, while hashed fingerprints like Daylight and ECFP employ hash functions to depict molecular patterns. Reaction formats like Reaction SMILES, RInChI, and CGR delineate reaction pathways, whereas macromolecules, including proteins and peptides, utilize sequence-based notations and structures sourced from repositories like the Protein Data Bank (PDB). This diverse array of methods facilitates accurate analysis and prediction within the realms of cheminformatics and drug discovery.
Graphical Representations for Molecules and Macromolecules
Graphical representations of molecules play a pivotal role in visualization and analysis, encompassing both 2D depictions and advanced 3D models. 2D depictions typically adhere to standardized guidelines set forth by the International Union of Pure and Applied Chemistry (IUPAC), albeit facing challenges in layout and rendering complexities. Tools such as RDKit and CDK have enhanced 2D visualizations, whereas for macromolecules, focus lies on depicting polymer or peptide structures, aided by specialized tools like the Pfizer Macromolecule Editor. 3D depictions, facilitated by software such as Avogadro and PyMOL, encompass diverse models such as ball-and-stick, cartoon, and van der Waals representations, thereby supporting studies in docking simulations, protein-ligand interactions, and mechanistic analyses. These graphical representations not only enhance understanding within the realms of cheminformatics and drug discovery but also underscore the interdisciplinary nature of modern computational techniques.
Conclusion:
The evolution of chemical representations and the integration of AI in drug discovery signify a transformative shift towards more efficient and scalable methodologies. This convergence enables accelerated data handling, advanced analytical capabilities, and enhanced visualization tools, positioning industries at the forefront of innovation in pharmaceutical research and development.