
- MIT introduces a groundbreaking method that merges first-principles calculations with machine learning.
- The method addresses the challenge of understanding thermal conductivity in semiconductors, particularly diamonds.
- It predicts strain hypersurfaces to effectively modulate diamond thermal conductivity through reversible elastic strain.
- Neural networks exploit structured correlations between band dispersion and strain for accurate predictions.
- Approximately 15,000 strain points are collected and analyzed using ab initio calculations.
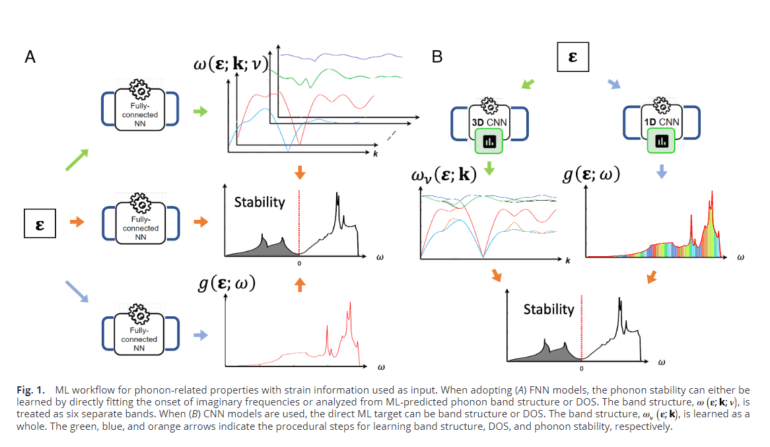
- Various machine learning models are trained to forecast phonon stability, density of states (DOS), and band structures for different strain states.
- Performance is enhanced through synergistic data sampling and active learning cycles.
- Molecular dynamics simulations validate computed thermal conductivity trends.
Main AI News:
MIT unveils a pioneering method merging first-principles calculations with machine learning, tackling the challenge of computationally intensive and formidable calculations essential for comprehending the thermal conductivity of semiconductors, with a specific emphasis on diamonds. Despite diamonds’ reputation as superb thermal conductors, comprehending the modulation of their lattice thermal conductivity via reversible elastic strain (ESE) remains a daunting task. This method aims to forecast the strain hypersurface where phonon instability arises, effectively altering the thermal conductivity of diamonds through profound ESE.
Historically, first-principles calculations have been pivotal in unraveling phonon band structure and associated characteristics. Nevertheless, these techniques are resource-intensive and may not be conducive to real-time computation. The proposed strategy involves harnessing neural networks to exploit the structured correlation between band dispersion and strain. To achieve accurate predictions of phonon stability, density of states (DOS), and band structures for strained diamond configurations, the researchers leverage data from ab initio calculations to train machine learning models.
The methodology commences with calibrating computational outcomes against experimental data for unaltered diamonds. Subsequently, approximately 15,000 strain points are amassed through Latin-Hypercube sampling and subjected to ab initio calculations to derive diverse properties for each distorted structure. Density functional theory (DFT) simulations are employed for structure relaxation, employing the Green-Lagrangian strain measure. Phonon computations are conducted based on density functional perturbation theory (DFPT). Various machine learning architectures, including fully connected neural networks and convolutional neural networks, are trained to forecast phonon stability, DOS, and band structures across different strain conditions.
The models’ performance is augmented through synergistic data sampling and active learning iterations. Moreover, molecular dynamics (MD) simulations are enlisted to compute a diamond’s thermal conductivity, offering qualitative validation of the identified trends.
To sum up, the research introduces an innovative approach to comprehending and adjusting the thermal conductivity of diamonds via reversible elastic strain. By harnessing machine learning models trained on first-principles calculations, the researchers can anticipate phonon stability and associated attributes for strained diamond configurations. This method provides a computationally efficient avenue to explore the intricate interplay between strain and thermal conductivity, unlocking possibilities for tailoring device performance and optimizing semiconductor figure-of-merit.
Conclusion:
This pioneering method from MIT represents a significant advancement in the understanding and manipulation of diamond thermal conductivity. Leveraging machine learning and first-principles calculations opens avenues for customizing semiconductor device performance, potentially reshaping the market landscape with optimized figure-of-merit in semiconductor technologies.