
TL;DR:
- Crystal structures profoundly impact material properties and research.
- Crystal structure prediction utilizes machine learning and graph theory to find stable structures.
- Machine learning reduces computation costs, while graph theory narrows the search space.
- Results show significant breakthroughs in various scientific fields.
Main AI News:
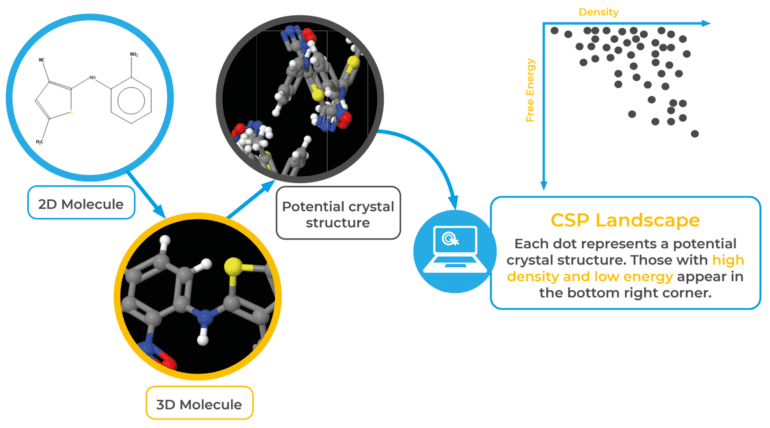
In the world of materials science, crystal structures play a pivotal role in determining the properties of materials. As such, research on crystal structures serves as the cornerstone for material studies. Crystal structure prediction, a method that seeks stable or metastable structures based solely on chemical composition under specific conditions, has emerged as a powerful tool for discovering novel materials and exploring uncharted phase space. With the ability to assist in experimental structure determination and guide synthesis, crystal structure prediction significantly reduces the burden of experimental costs, making it a matter of great scientific importance.
Yet, existing crystal structure prediction techniques face formidable limitations stemming from two critical aspects. Firstly, the cost of performing structure optimization through first-principles calculations escalates cubically with the number of atoms involved. Secondly, the number of local minima on the potential energy surface multiplies exponentially with increasing degrees of freedom. Such hindrances have impeded progress in this crucial domain of materials science.
To confront the first challenge head-on, a groundbreaking article proposes the integration of machine learning potentials for surrogate calculations. The process commences with the generation of random structures, from which a subset is selected randomly to undergo Density Functional Theory (DFT) single-point energy, force, and stress calculations, creating a comprehensive training set. The machine learning potential is then meticulously trained using this dataset. As the subsequent search process unfolds, the computationally intensive DFT structure optimizations are seamlessly replaced with the efficient machine learning force field. During the optimization journey, structures necessitating extrapolation are diligently noted. If the extrapolation exceeds a predetermined threshold, DFT single-point calculations are promptly executed on these structures, subsequently incorporating them into the training set for retraining the force field, fine-tuning the potential function. This iterative process persists until the optimization can be flawlessly conducted by the machine learning surrogate. To compensate for any errors introduced by the machine learning force field, calibration using first-principles calculations is essential upon the search’s conclusion.
Addressing the second challenge, the research team embraced the power of graph theory to reduce the vast search space. In evolutionary algorithms, preserving exceptional “genes” from the parent generation to the offspring generation is a pivotal concept. In the context of crystal structures, these “genes” correspond to local structural motifs. While identifying such motifs is relatively straightforward in molecular crystal searches, it poses a considerable challenge for ordinary extended crystals. However, by abstracting periodic crystal structures into graphs, community detection algorithms come to the rescue. For instance, consider the extended α-boron structure; when represented as a quotient graph and subjected to community detection algorithms, the boron icosahedron emerges as the extracted 0-dimensional component. By conserving this component throughout subsequent genetic operations, the degrees of freedom are dramatically reduced from 36 to a mere 6, resulting in a significant reduction of the overall search space.
The research team’s efforts bore remarkable fruits during subsequent testing. The machine learning approach impressively slashed the number of self-consistent calculations by nearly two orders of magnitude. Simultaneously, the ingenious application of graph theory facilitated a 30% reduction in the number of structures that needed exploration to reach the target structure. These impressive results have opened up exciting new avenues in fields such as planetary science, superhard materials, high-energy density materials, and superconducting materials, propelling the realm of crystal structure predictions into a new era of possibilities.
Conclusion:
The integration of machine learning and graph theory in crystal structure predictions offers a transformative approach to materials research. The application of these advanced techniques not only reduces computational expenses but also expedites the discovery of new materials, making it a game-changer for the market of materials science and engineering. With the potential to unlock groundbreaking possibilities in diverse sectors, from planetary science to high-energy density materials, businesses and industries will witness a surge in innovation and efficiency, driving the market towards unprecedented growth and advancement.