
TL;DR:
- Microsoft AI Research introduces DiG, a novel deep learning framework for equilibrium distribution-based protein structure prediction.
- DiG enables modeling ensembles of structures based on equilibrium distributions, offering a more comprehensive understanding of molecular systems.
- The framework utilizes deep neural networks and fundamental molecular descriptors to directly forecast target distributions.
- Inspired by simulated annealing, DiG gradually refines simple distributions to build complex distributions, mimicking the annealing process.
- DiG demonstrates efficient and affordable generation of realistic molecular structures and provides estimates of state densities.
- This breakthrough represents a significant advancement in quantitatively analyzing microscopic molecules and predicting their macroscopic features.
Main AI News:
Molecular science faces a significant challenge in accurately predicting the structure and properties of molecules. Recognizing the importance of structure prediction, scientists have embraced deep learning approaches such as AlphaFold and RoseTTAFold, which have achieved remarkable accuracy in identifying protein structures based on their amino acid sequences. However, these methods offer only a glimpse into a protein’s function, providing a singular snapshot of its structure.
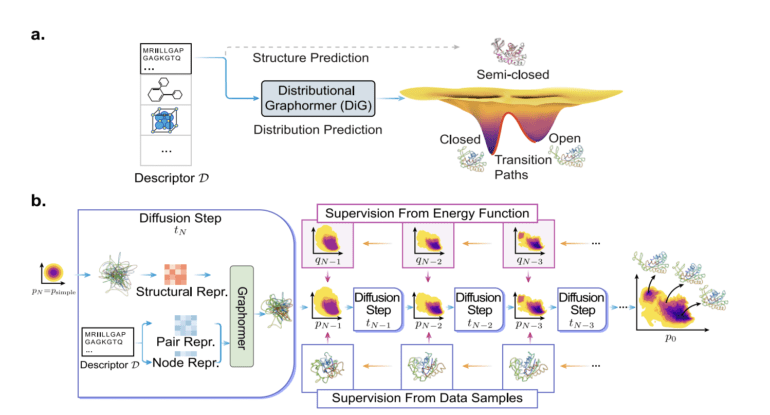
In a groundbreaking development, Microsoft AI Research has unveiled the Distributional Graphormer (DiG), a novel deep learning framework designed to address the limitations of traditional structural prediction methods. DiG represents a major leap forward in the field of equilibrium distribution-based protein structure prediction, aiming to revolutionize molecular science. By modeling ensembles of structures based on equilibrium distributions rather than relying on a single structure, DiG opens new doors for applying statistical mechanics and thermodynamics at both the microscopic and macroscopic levels of molecular systems.
DiG builds upon the success of its predecessor, Graphormer, a versatile graph transformer capable of accurately describing molecular structures. This enhanced version, DiG, introduces a fresh approach to distribution prediction, leveraging the power of deep neural networks and fundamental molecular descriptors to directly forecast target distributions. The utilization of simulated annealing, a proven technique in thermodynamics and optimization, serves as the foundation for DiG’s concept. Simulated annealing has already propelled advancements in the realm of artificially generated content (AIGC) and diffusion models, enriching the field in recent years.
Drawing inspiration from annealing processes, DiG refines a simple distribution into a complex one by progressively exploring and settling in the most probable states. Acting as a deep learning framework for molecular systems, DiG mimics this annealing procedure. Diffusion models, rooted in statistical mechanics and thermodynamics, frequently serve as the basis for AIGC models, reinforcing the scientific underpinnings of DiG’s methodology.
DiG employs Graphormer’s capability to transform a simple distribution into a complex one through diffusion. The training data used by DiG can be flexible, as it minimizes the disparity between energy-based probabilities and those predicted by DiG. By utilizing the energy functions of molecular systems, DiG can guide the transformation process. Leveraging existing knowledge, DiG is primed to learn and adapt.
To demonstrate the efficacy and potential of DiG, the Microsoft research team conducted a series of molecular sampling tasks encompassing diverse molecular systems, including proteins, protein-ligand complexes, and catalyst-adsorbate systems. The results showcase DiG’s ability to generate realistic and diverse molecular structures efficiently and affordably. Furthermore, DiG provides estimates of state densities, which are critical for computing macroscopic attributes using statistical mechanics.
Conclusion:
The introduction of DiG signifies a major breakthrough in molecular science. By revolutionizing the prediction of protein structures and enabling a deeper understanding of molecular systems, DiG opens up new avenues for research and exploration. Its ability to efficiently generate realistic molecular structures and estimate state densities has the potential to transform various industries reliant on molecular analysis, such as pharmaceuticals, materials science, and biotechnology. The market can expect accelerated advancements and novel applications in these fields as scientists leverage DiG’s capabilities for innovation and discovery.