
- Syntheseus, a new Python library, simplifies the evaluation of machine learning in retrosynthetic planning.
- Developed by Microsoft researchers and collaborators, it integrates eight reaction models into a unified interface.
- Addresses challenges of assessing synthesizability and standardizes evaluation metrics.
- Enables comprehensive assessment from input molecule to synthesis pathway.
- Supports retraining with larger datasets and comes with default weights trained on USPTO-50K.
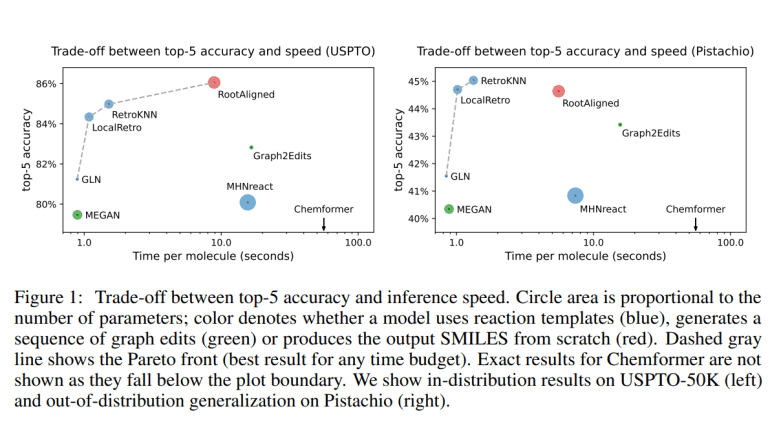
- Evaluation highlights RetroKNN as a top-performing model across various criteria.
- Holds promise for advancing multi-step retrosynthesis planning.
Main AI News:
The convergence of chemistry and machine learning has captured considerable attention in recent times. However, the application of advanced reaction models poses significant hurdles. These models, known for their complexity, lack easily accessible entry points, complicating their utilization beyond benchmarking. To address this, researchers from Microsoft, along with collaborators from esteemed institutions, have developed Syntheseus—a Python library aimed at streamlining the evaluation of retrosynthesis algorithms.
Syntheseus addresses the challenge of assessing the synthesizability of generated molecules by providing a comprehensive platform for benchmarking. By integrating eight free and open-source reaction models into a unified interface, Syntheseus simplifies the comparison of various methodologies. Gone are the days of grappling with diverse codebases; now, researchers can seamlessly evaluate different models using a standardized approach.
In their endeavor to establish best practices, the team scrutinized existing metrics used in retrosynthesis evaluation. By analyzing previous studies, they seek to provide clarity on the efficacy of retrosynthesis algorithms. Syntheseus not only facilitates evaluation but also encourages consistency, ensuring that assessments are conducted rigorously across different methodologies.
One of the key challenges in retrosynthesis evaluation is the balance between experimental validation and computational efficiency. Synthesizing molecules in the lab is resource-intensive and time-consuming, making it impractical for algorithm development. Moreover, existing studies often focus on single-step retrosynthesis, overlooking the holistic process. Syntheseus addresses these limitations by offering a platform for comprehensive evaluation, from initial molecule input to synthesis pathway generation.
By leveraging datasets like USPTO-50K and Pistachio, Syntheseus enables researchers to evaluate model performance across diverse chemical reactions. The library comes equipped with default weights trained on USPTO-50K, eliminating the need for researchers to hunt for model parameters. Additionally, Syntheseus supports retraining using larger datasets, allowing for continual improvement of models.
The evaluation conducted by the researchers sheds light on the strengths and limitations of existing models. While some models excel in specific metrics, others demonstrate superior performance in broader contexts. Notably, RetroKNN emerges as a frontrunner across various evaluation criteria, showcasing its versatility and efficiency.
Looking ahead, Syntheseus holds promise for advancing multi-step retrosynthesis planning. Although preliminary results are presented, the framework developed by the researchers lays the groundwork for optimizing end-to-end synthesis pipelines. As the field continues to evolve, Syntheseus will play a pivotal role in shaping the future of molecular design and synthesis.
Conclusion:
The introduction of Syntheseus marks a significant step forward in the integration of machine learning into retrosynthetic planning. By providing a unified platform for evaluation and benchmarking, it streamlines the development and assessment of chemical synthesis algorithms. This advancement is poised to accelerate innovation in the chemical industry, driving efficiencies in molecule design and synthesis processes. As Syntheseus continues to evolve, it will likely shape the landscape of molecular design software and contribute to the ongoing convergence of chemistry and artificial intelligence.