
TL;DR:
- MIT researchers explore the scalability of deep learning models in chemistry research.
- Focus on generative pre-trained transformers (ChemGPT) and graph neural network force fields (GNNs).
- Introduction of the “neural scaling” concept to characterize model performance.
- The study examines power-law relationships in loss scaling concerning model parameters, dataset size, and compute resources.
- Design and pre-train ChemGPT using SELFIES representations of molecules from PubChem.
- Investigation into the impact of dataset size and model dimensions on pre-training loss.
- Evaluation of four types of GNNs for molecular geometry and structure tasks.
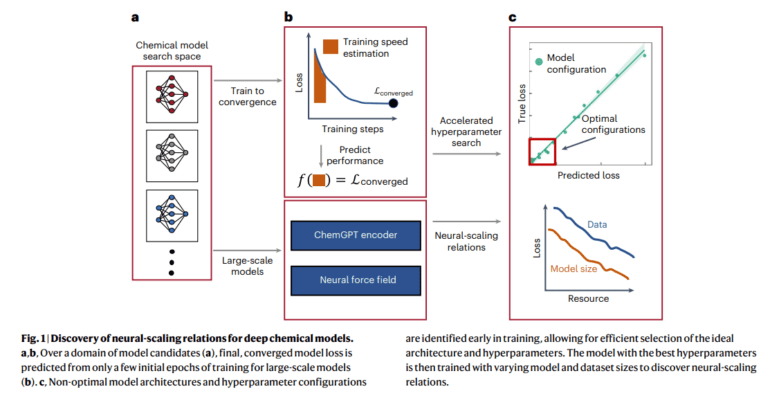
- Introduction of Training Performance Estimation (TPE) for hyperparameter optimization in deep chemical models.
- Use of NVIDIA Volta V100 GPUs, PyTorch, and distributed data-parallel acceleration in experiments.
- Insights contribute to resource-efficient model architectures in scientific deep learning.
Main AI News:
In this compelling study conducted by researchers at MIT, an in-depth investigation into the scalability of deep learning models within the field of chemistry research is unveiled. This research specifically focuses on the application of generative pre-trained transformers (GPT) tailored for chemistry, known as ChemGPT, and the utilization of graph neural network force fields (GNNs). The central concept introduced here is that of “neural scaling,” a pivotal notion that characterizes model performance through empirical scaling laws.
The study places emphasis on the scaling of these models concerning various factors, such as the number of model parameters, dataset size, and computational resources, viewing these aspects as power laws in relation to loss scaling. By delving into this uncharted territory, the researchers seek to shed light on the challenges and opportunities that arise when scaling up large chemical models, ultimately aiming to offer valuable insights into the optimal allocation of resources for enhancing pre-training loss.
For the realm of chemical language modeling, the researchers have ingeniously devised ChemGPT—a model styled after the formidable GPT-3 but tailored for chemical applications, utilizing a tokenizer capable of handling self-referencing embedded strings (SELFIES) to represent molecules. This sophisticated model undergoes pre-training using a dataset sourced from PubChem, and the study meticulously dissects the impact of both dataset size and model dimensions on pre-training loss, ensuring a thorough examination of the interplay between these variables.
In addition to their exploration of language models, the paper also delves into the realm of graph neural network force fields (GNNs), a crucial tool for tasks demanding a deep understanding of molecular geometry and three-dimensional structures. Four distinct types of GNNs are under scrutiny, encompassing models with internal layers exclusively manipulating E(3) invariant quantities to more complex architectures incorporating E(3) equivariant quantities with a physics-informed approach. The authors evaluate the capacity of these GNNs, meticulously considering both depth and width during their neural-scaling experiments.
The research further introduces an innovative technique named Training Performance Estimation (TPE), initially adapted from methodologies commonly employed in computer vision architectures. TPE serves as an efficient means to tackle hyperparameter optimization (HPO) for deep chemical models. This ingenious method harnesses training speed to facilitate performance estimation across diverse domains and varying model and dataset sizes. The paper takes a detailed look at the experimental setup, highlighting the utilization of NVIDIA Volta V100 GPUs, PyTorch, and distributed data-parallel acceleration for the implementation and training of these intricate models.
Conclusion:
MIT’s groundbreaking research on neural scaling in chemistry opens up new avenues for the efficient utilization of deep learning models in scientific applications. This exploration provides valuable insights into resource-efficient model architectures, which can revolutionize the market by enhancing the capabilities of chemical research and development, ultimately driving innovation and efficiency in the industry.