
TL;DR:
- ADMET-AI, developed by Stanford researchers, combines AI and chemical databases for drug discovery.
- It addresses the challenge of identifying drug candidates with ideal properties.
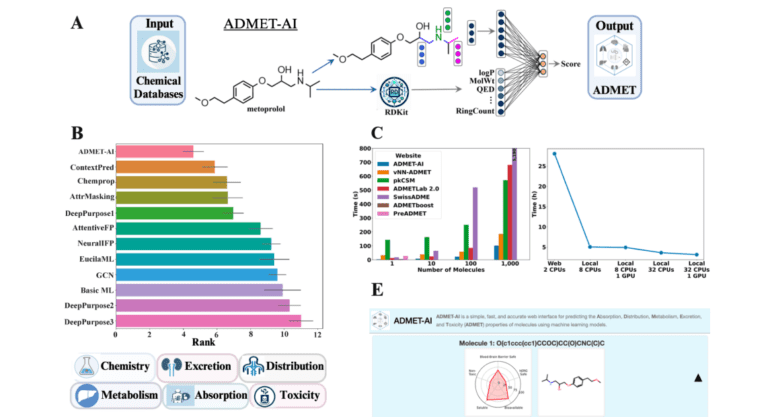
- It utilizes Chemprop-RDKit, a graph neural network trained on 41 datasets for accuracy and speed.
- Unique features include batch predictions and contextualized forecasts based on approved drugs.
- ADMET-AI’s architecture integrates 200 molecular features, outperforming competitors.
- Demonstrated excellence across 41 ADMET datasets with high-speed web server and local capabilities.
Main AI News:
The fusion of expansive chemical databases with the integration of generative AI into the realm of drug discovery has ushered in a promising era of drug candidate exploration. Yet, the ultimate challenge lies in the precise identification of compounds possessing optimal druglike attributes, particularly those pertaining to absorption, distribution, metabolism, extraction, and toxicity (ADMET). Traditional screening approaches, with their inherent tedium, often fall short of delivering the level of accuracy required for this critical task. In response to this challenge, a collaboration between Stanford University and Greenstone Biosciences has given rise to ADMET-AI, an advanced machine-learning platform poised to forecast ADMET properties swiftly and reliably for extensive chemical libraries.
In the landscape of drug discovery, high-throughput docking and generative AI methodologies have significantly expanded the pool of potential drug candidates. However, these methods occasionally yield molecules that may not align with the stringent criteria for effective pharmaceutical agents. This underscores the pressing need for a screening tool that embodies both speed and precision. Enter ADMET-AI, a visionary solution that harnesses the power of Chemprop-RDKit, a graph neural network. This exceptional network has been meticulously trained on 41 datasets sourced from the Therapeutics Data Commons, endowing it with the capability to outshine its counterparts in terms of both speed and accuracy. ADMET-AI boasts distinctive features, including the ability to make predictions on batches of molecules and provide contextually informed forecasts based on a repository of approved drugs.
At the core of ADMET-AI’s architecture lies the remarkable integration of Chemprop-RDKit, a fusion of a graph neural network with 200 physicochemical molecular features computed by RDKit. This unique amalgamation empowers the model to make highly accurate predictions across a wide spectrum of ADMET properties, an achievement that has propelled it to attain the highest average rank on the TDC ADMET Benchmark Group leaderboard. The platform’s efficacy shines brightly across 41 TDC ADMET datasets, showcasing unparalleled performance in both regression and classification tasks. Notably, the web server’s speed is nothing short of extraordinary, clocking in at a remarkable 45% faster than its closest ADMET web server competitor. Furthermore, the local version of ADMET-AI adds a practical dimension by delivering high-throughput prediction capabilities, processing a staggering one million molecules in a mere 3.1 hours.
Conclusion:
ADMET-AI’s emergence as a transformative force in drug discovery signifies a shift towards precision and speed in identifying potential drug candidates. Its ability to swiftly and accurately predict ADMET properties, combined with unique features and outstanding performance, is set to reshape the pharmaceutical research and development landscape, offering new opportunities and efficiencies in the market.