
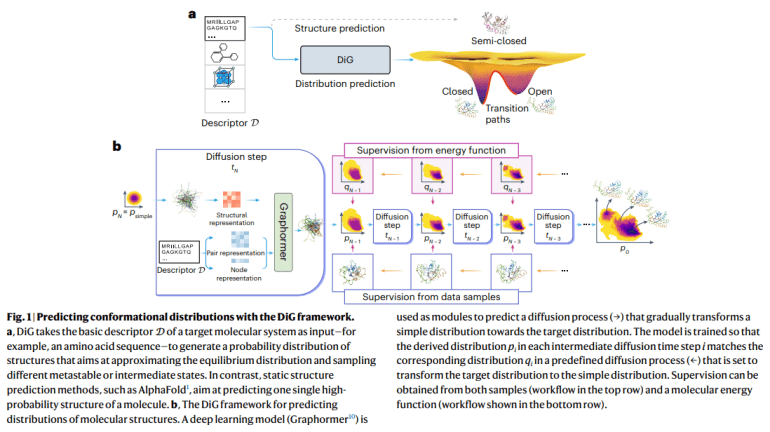
- DiG, a deep learning framework, revolutionizes equilibrium distribution prediction in molecular systems.
- Inspired by thermodynamic principles, DiG employs neural networks to transform simple distributions into complex equilibrium states.
- Through a diffusion process, DiG efficiently generates diverse molecular conformations, enabling property-guided structure generation.
- DiG showcases versatility across various molecular tasks, from protein conformation sampling to ligand structure prediction.
- Its accuracy and efficiency position DiG as a frontrunner in molecular dynamics, promising transformative discoveries in drug design and materials science.
Main AI News:
In the ever-evolving field of molecular dynamics, a groundbreaking solution emerges to tackle the intricacies of equilibrium distribution prediction. DiG, born from the collaborative efforts of researchers spanning the globe, heralds a new dawn in deep learning applications tailored for molecular systems. Building upon its foundation inspired by thermodynamic principles, DiG represents a paradigm shift towards efficient and accurate equilibrium distribution prediction.
At its core, DiG leverages the power of neural networks to traverse the vast landscape of molecular structures, transforming simple distributions into complex equilibrium states. Through a meticulous diffusion process, DiG unravels the mysteries of molecular flexibility, enabling the generation of diverse conformations with unparalleled speed and precision. This transformative approach not only revolutionizes molecular modeling but also opens doors to property-guided structure generation, empowering researchers to design structures with desired properties.
The versatility of DiG extends far beyond prediction, offering solutions to an array of molecular challenges. From protein conformation sampling to ligand structure prediction, DiG demonstrates its adaptability and accuracy, setting new standards in the field. Its prowess in catalyst-adsorbate sampling further cements its position as a frontrunner in molecular dynamics, providing researchers with invaluable insights into catalytic processes.
In conclusion, DiG stands as a beacon of innovation in the realm of molecular dynamics, promising to reshape the landscape of scientific inquiry. With its ability to predict equilibrium distributions and facilitate property-guided design, DiG paves the way for transformative discoveries in drug design, materials science, and beyond. As we embark on this journey into the future of molecular dynamics, DiG lights the path towards unprecedented advancements and discoveries.
Conclusion:
The emergence of DiG marks a significant leap forward in molecular dynamics, offering unparalleled capabilities in predicting equilibrium distributions and guiding structure generation. This innovation has the potential to revolutionize the market by accelerating drug design processes, enhancing materials discovery, and opening new avenues for scientific inquiry and innovation. Businesses operating in these sectors should closely monitor the development and adoption of DiG to stay competitive in a rapidly evolving landscape.